Methods
DDGun is an untrained method for predicting the cariation of unfolding free energy changeg upon mutation (ΔΔG). DDGun is an algorithm based on evolutionary information which predicts the unfolding ΔΔG for single and multiple variations. The predictions are performed through a linear combination of scores derived from protein sequence and structural features. The three following scores are based purely on sequence data:
the difference between the wild type and mutant residue in the Blosum62 substitution matrix (sBl);
the difference in the interaction energy (Skolnick statistical potential) between the wild-type and substituted residue with their sequence neighbours within a 2-residue window (sSk);
the difference in the hydrophobicity between wild type and mutant residues according to the Kyte-Doolittle scale (sHp).
We also developed, a structure-based version of DDGun (DDGun3D) adding two structure-based terms in the input features. The first structural term represents the
difference in the interaction energy (Bastolla statistical potential) between the wildtype and mutant residue with its structural neighbours (sBV).
The second structural term is the relative solvent accessibility of the residue (ac), computed as the current accessibility divided by its maximum value.
The first four scores are linearly combined while the latter is used to modulate the mutation effect with the residue accessibility. This effect is obtained by
multiplying the total score by (1-ac). For a better tuning of the predictions of also fully accessible residues (ac = 1), the modulation factor
was set to (1- ac + ε), where ε was arbitrarily set to 0.1.
All first four scores described above were weighted through the profile built on the multiple sequence alignment of the protein and its homologues.
The multiple sequence alignement is built using the hhblits program from the hh-suite
running on the UniRef30 database (uniclust30_2018_08).
In detail the weighted scores are caluclated using the following equations:
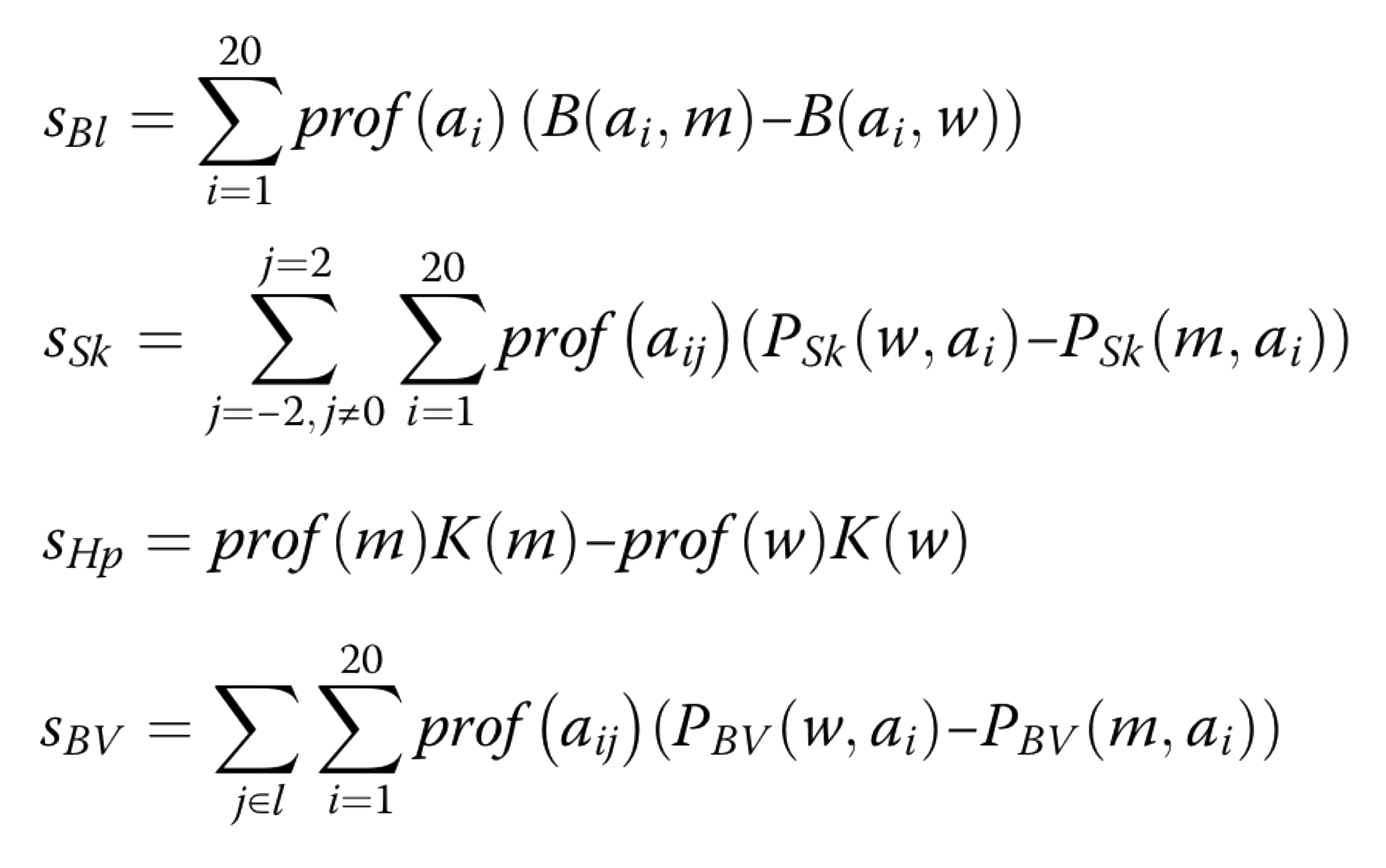
The scores described above are combining using different weights for the sequence ad structure based methods. In the equantions below are reported the coefficients
of each score for the sequence-based (sseq) structure-based (s3DA) methods:
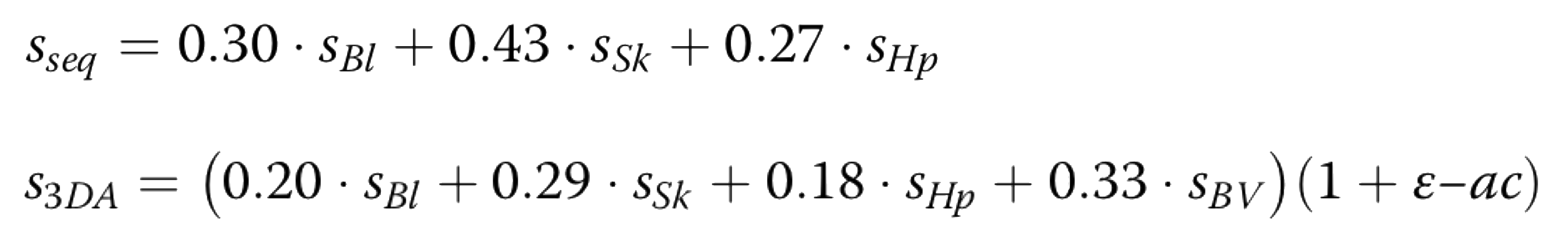
This method can be adapted to the prediction of the ΔΔG for multiple site variations.
Indeed for each multiple-site variation we compute the score for each single site variation comprising it.
Given a multiple site variation with multiplicity M (that is composed of M single site variations), let name ss
the vector of M single site scores; ss = (s1, s2, …sM).
We compute the score for a multiple site variants as:
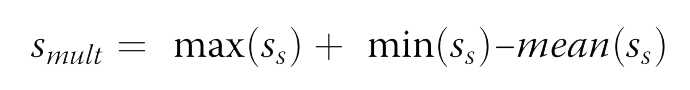
For single point variations, the following data sets were considered: the most commonly used S2648; the high quality VariBench which was integrated with the 605
manually curated variations selected in Broom et al. for a total of 1900 high quality variations; a data set of variations on the P53 protein and myoglobin data sets.
The dataset for multiple site variations was derived from ProTherm. A total of 914 protein multiple site variations, with a number of simultaneous variants
ranging from 2 to 10, were derived. We called this set of multiple site variations PTmul.
The performance of DDGun and DDGun3D were tested on different datasets calculating the Pearson correlation coefficient
(PCC) and the Root Mean Square Error
(RMSE)
between the experimental and predicted ΔΔG values. In the table below we summarized the performance of DDGun and DDGun3D on different datasets of single
(VariBench, S2648, Ssym ) and multiple (PTmul) site protein variants reporting for each of them the PCC and RMSE values.
The performance of both methods are compared with those achived by selected group of algorithms. For this comparison we calculated the performance of all
the methods on two datasets of single (s96) and multiple (m28) site amino acid variants. The results of this comparison is reported in the following table.
All the predictions and datasets are available in the tar file ddgun-preds.tar.gz. The following repository on zenodo includes all the predictions calculated with in first version of DDGun which run hhblits on the uniprot20_2016_02 database.